
Interesting Case February 2023
Case contributed by:
Paula Toro Castano, MD, PGY-2 Pathology Resident, and Clifton Fulmer, MD, PhD, Staff Gastrointestinal and hepatobiliary pathologist
Cleveland Clinic
Have an interesting case to share?
Contact HPHS newsletter committee chair Sadhna.Dhingra@ProPath.com
Case history
The patient was born at 39 weeks gestational age via spontaneous vaginal delivery. Induction of labor was required for unstable fetal lie. After delivery, the patient was transferred to the neonatal intensive care unit for hypoglycemia. Importantly, their bilirubin peaked at 8 mg/dL. The patient was discharged to home on hospital day 17.
Two weeks after being discharged (1 month and 1 week of age), the patient presented to the emergency department with jaundice, direct bilirubin of 3.9 mg/dL (normal range <2 mg/dL), and a GGT of 646 U/L (normal range 6-46 U/L). An abdominal ultrasound showed a small irregular gallbladder without visualization of the common biliary duct and was concerning for biliary atresia (BA). The patient was admitted to the hospital for further evaluation. Additional laboratory workup during that admission revealed an MMP-7 of 22.5 ng/ml (IQR: 8.54-24.55 ng/ml for non-BA), indicating a low probability of BA, ammonia within normal limits, indirect antiglobulin negative, CMV negative, Alpha 1 antitrypsin (A1AT) of 28 mg/dL (normal range 100-220 mg/dL), and TSH normal. The patient had a liver biopsy performed 1 week following readmission, which was significant for a paucity of bile ducts and hepatocellular cholestasis. A PAS stain was negative for abnormal globular deposits within periportal hepatocytes. A HIDA scan did not demonstrate a definitive diagnosis of biliary atresia. However, biliary excretion of tracer was abnormal with no clearance from the liver or gallbladder/bowel activity during imaging at both 1-hour and 24 hour time points. Given this abnormal imaging and the clinical concern for BA, the patient was discharged and scheduled for a cholangiogram and A1AT genetic testing.
The patient was readmitted for scheduled cholangiogram. Their A1AT phenotype returned abnormal (PiZZ consistent with A1AT deficiency). The cholangiogram was concerning type IIa BA. The patient subsequently underwent Kasai hepaticoportoenterostomy at 1.5 months of age. The procedure was complicated by portal hypertension, ascites, and growth failure requiring living-related donor liver transplantation that was performed at 10 months of age.
A week after transplantation, the patient was readmitted for low-grade fever and decreased energy level requiring a 2-day hospitalization.
Fortunately, the patient continues to do well since their last clinic visit.
Pathology findings
Histologic sections of the explanted liver show nodular remodeling of hepatic parenchyma by thick fibrous bands and septae that is characteristic of established cirrhosis. Irregular, “jigsaw puzzle”-like regenerative nodules are also present, imparting a biliary appearance to the cirrhotic liver (Figures 1 and 2). The identifiable portal tracts and fibrous septae contain a sparse, predominantly lymphohistiocytic inflammatory infiltrate. There is no significant interface activity and plasma cells are inconspicuous. While a minority of the smallest portal tracts contain an identifiable native bile duct branch, most do not (Figure 3). In fact, none of the largest portal tracts contain a bile duct branch (Figures 4). There is, however, a brisk proliferation of bile ductules (Figure 5) at the interface of all of the portal tracts and fibrous septae with the adjacent lobular parenchyma. There is portal edema associated with this ductular reaction, as well as more than occasional polymorphonuclear inflammatory cells. Some bile ductules contain inspissated bile. The lobular parenchyma is relatively bland overall. A minority of zone 1, periportal hepatocytes contain macrovesicular fat droplets (Figure 6). There are rare foci of canalicular cholestasis, though this is mild overall. Lobular inflammation is minimal. An iron special stain shows patchy, trace hepatocellular iron (0 to 1+). The trichrome stain highlights the fibrous bands and septae surrounding the regenerative nodules. A PAS/D special stain highlights numerous conspicuous periportal/perisepatal cytoplasmic globules (Figure 7).
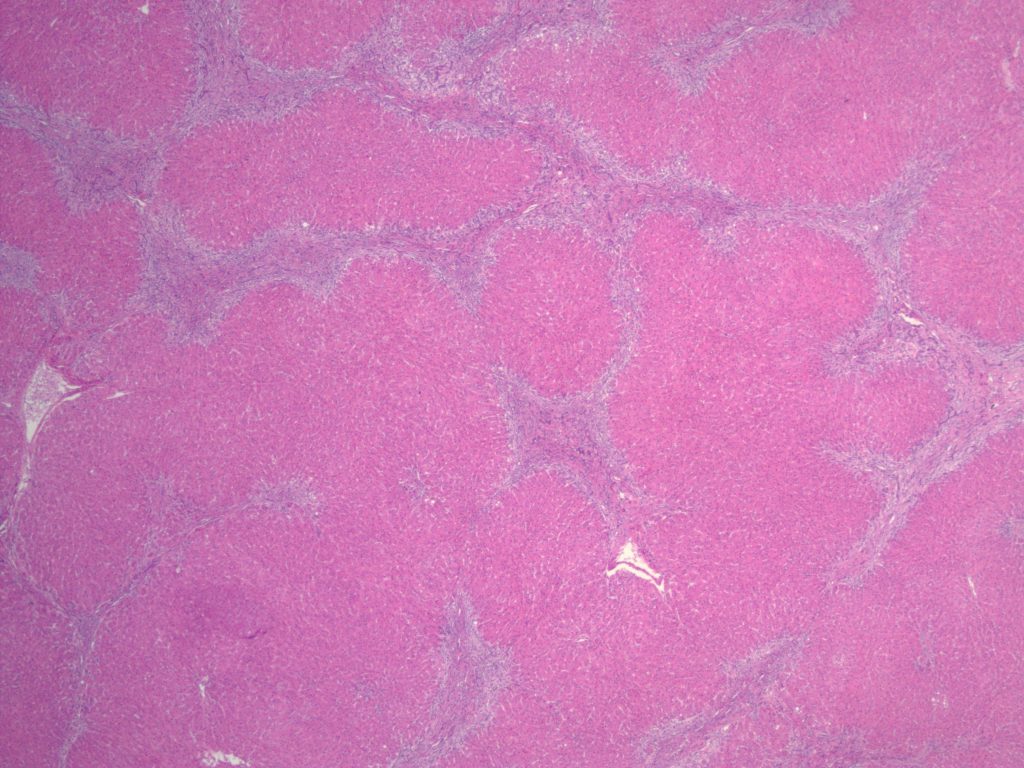
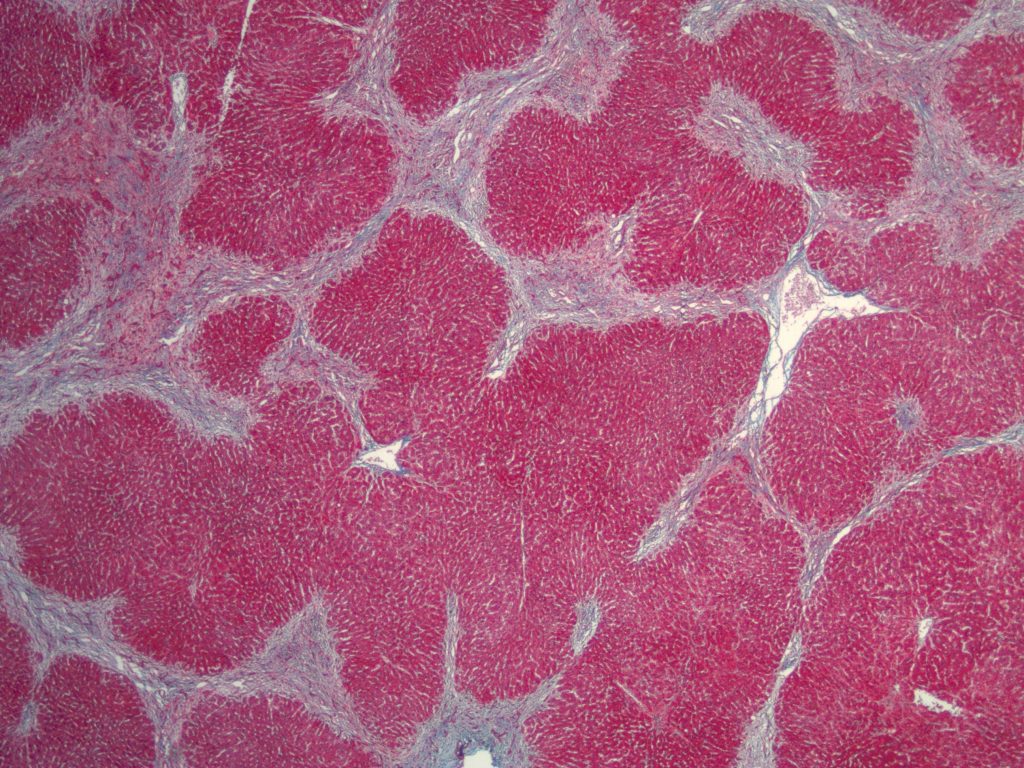
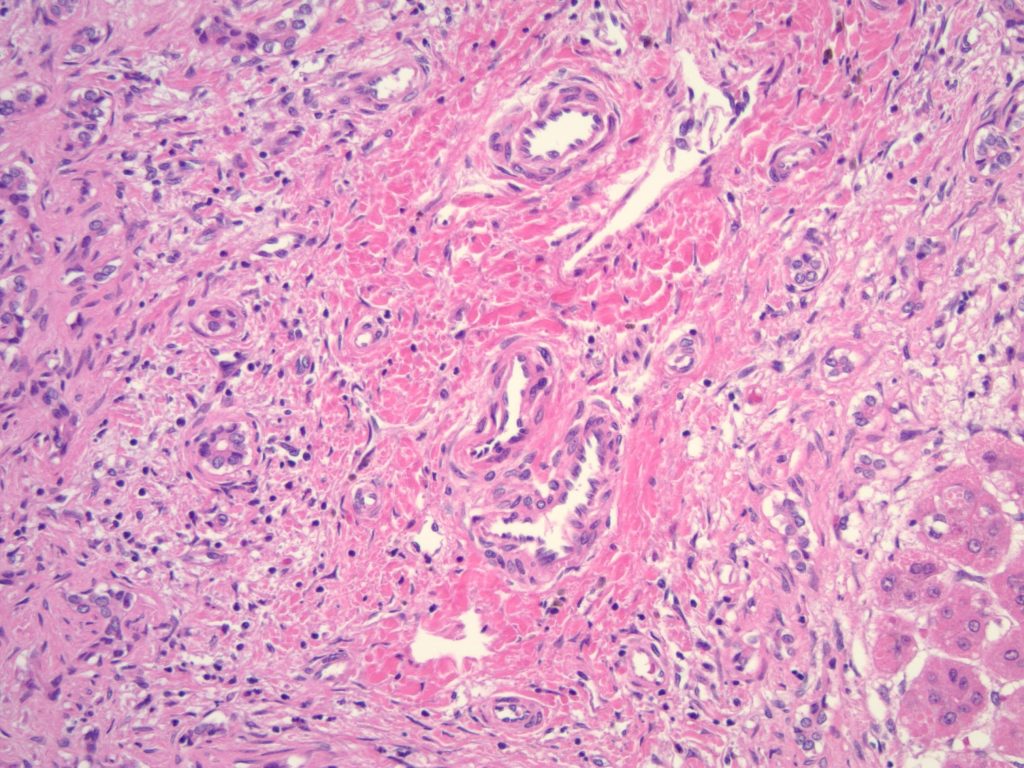
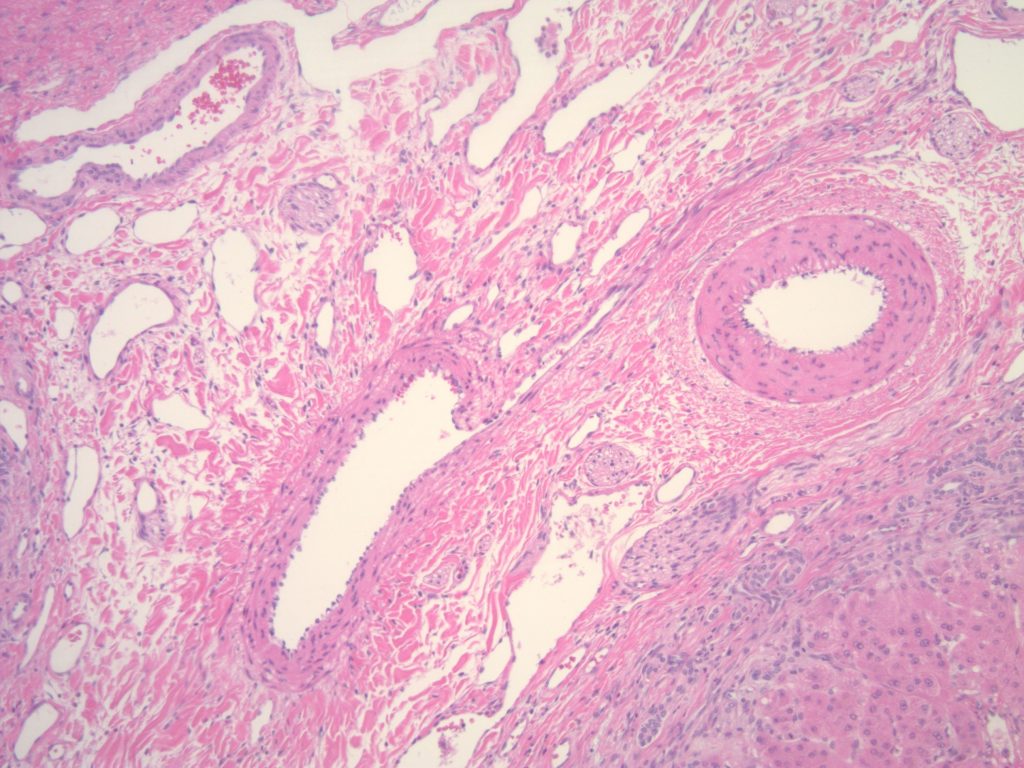
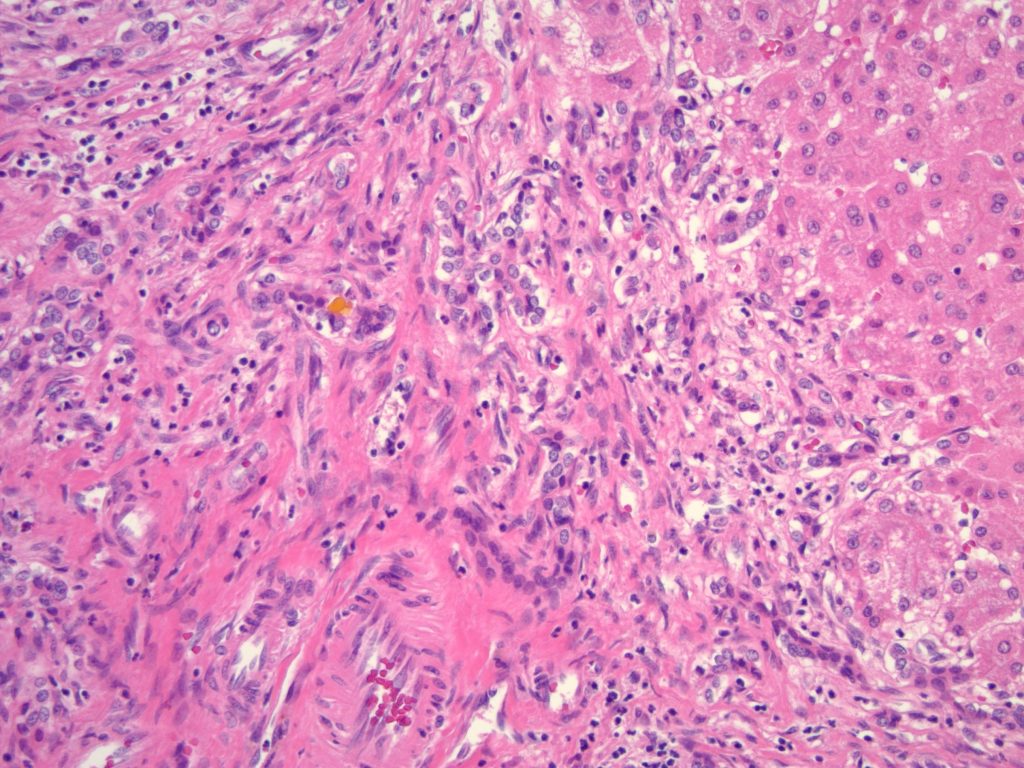
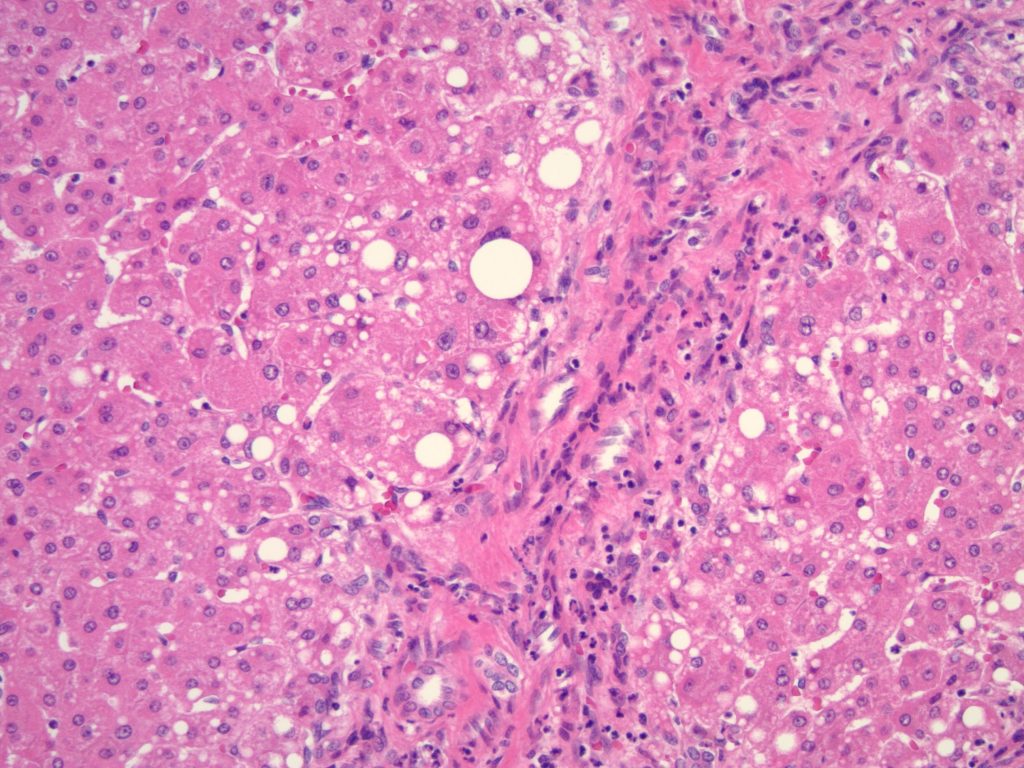
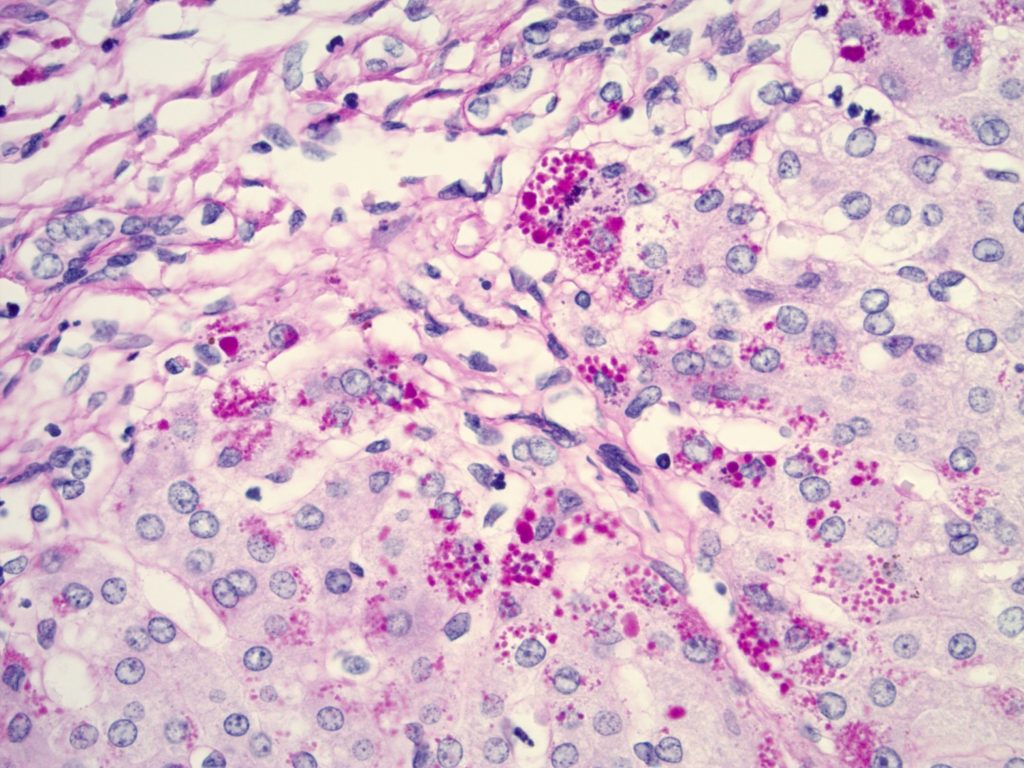
Diagnosis
Secondary biliary cirrhosis with marked ductopenia developing in the setting of alpha-1-antitrypsin deficiency (PiZZ), status post portoenterostomy (Kasai procedure).
Discussion
Alpha1-Antitrypsin deficiency (A1ATD) is a metabolic, hereditary disorder that is characterized by a low serum concentration of A1AT, a protease inhibitor. Patients with A1ATdeficiency have an imbalance in protease-antiprotease activity which corresponds to the main pathogenic factor of the disease (1). This entity is the most common genetic cause of pediatric liver disease and transplantation, and it must be considered when evaluating cholestatic infants by performing early molecular genetic testing (2).
PiZZ and PiSZ are the most common phenotypes and are associated with the greatest pathogenicity. Those who are homozygous for PiZZ can also present with liver disease due to severe protein misfolding and, as A1AT is secreted from hepatocytes, causing the accumulation of polymerized protein and hepatocellular damage. There is a bimodal pattern of clinical presentation with liver injury manifesting either during the neonatal period as cholestasis or acute liver failure, or in adulthood where chronic hepatocyte destruction eventually manifests with the development of fibrosis, and eventually established cirrhosis (3).
Histologically, A1T1D is classically characterized by the presence of PAS-positive, diastase-resistant globules within the cytoplasm of periportal hepatocytes. This may be accompanied by fibrosis that can range from minimal to established cirrhosis, as well as with mild chronic portal inflammation and periportal steatosis (4). However, in neonates, the classic intracytoplasmic globules may be difficult to detect or entirely absent, particularly in individuals less than 12 weeks of age. Additionally, the neonatal population can also present with extrahepatic BA-like features including lobular cholestasis, bile ductular reaction, giant cell transformation of hepatocytes, and variable fibrosis (5). These BA-like features could be misdiagnosed as BA if the patient’s A1AT phenotype is not known (6).
In this case, both A1ATD and BA were diagnosed. However, multiple studies have demonstrated that the identification of an alternative definitive diagnosis makes an unequivocal diagnosis of BA unlikely. Identification of an alternative definitive diagnosis such as A1ATD, cystic fibrosis, or Alagille syndrome is essential as this may identify a population of patients that are unlikely to benefit from a Kasai hepatoportoenterostomy (7).
Although not necessary for all infants with neonatal cholestasis, surgical exploration with operative cholangiography and pathologic examination of a bile duct remnant remains the only definitive means of making the diagnosis of biliary atresia (7) and should be accompanied by genetic testing for A1ATD.
Learning points:
- Molecular studies for A1T1 deficiency are recommended in cholestatic neonates
- Biliary atresia-like features are part of the spectrum of findings in patients with A1T1 deficiency
- Biliary atresia is unlikely if additional diagnostic entities such as cystic fibrosis, alpha-1 antitrypsin deficiency, and Alagille syndrome are identified
- Patients with A1ATD and BA-like features are unlikely to benefit from a Kasai hepatoportoenterostomy.
References
- Torres-Durán M, Lopez-Campos JL, Barrecheguren M, Miravitlles M, Martinez-Delgado B, Castillo S, et al. Alpha-1 antitrypsin deficiency: outstanding questions and future directions. Orphanet J Rare Dis. 2018 Dec;13(1):114.
- Khan Z, Venkat VL, Soltys KA, Stolz DB, Ranganathan S. A Challenging Case of Severe Infantile Cholestasis in Alpha-1 Antitrypsin Deficiency. Pediatr Dev Pathol. 2017 Apr;20(2):176–81.
- Brantly M, Campos M, Davis AM, D’Armiento J, Goodman K, Hanna K, et al. Detection of alpha-1 antitrypsin deficiency: the past, present and future. Orphanet J Rare Dis. 2020 Dec;15(1):96.
- Townsend SA, Edgar RG, Ellis PR, Kantas D, Newsome PN, Turner AM. Systematic review: the natural history of alpha-1 antitrypsin deficiency, and associated liver disease. Aliment Pharmacol Ther. 2018 Apr;47(7):877–85.
- Alpha1-antitrypsin Granules in the Liver–Always Important? QJM Int J Med [Internet]. 1990 Jul [cited 2022 Jun 5]; Available from: https://academic.oup.com/qjmed/article/76/1/699/1634977/Alpha1antitrypsin-Granules-in-the-LiverAlways
- Russo P, Magee JC, Anders RA, Bove KE, Chung C, Cummings OW, et al. Key Histopathologic Features of Liver Biopsies That Distinguish Biliary Atresia From Other Causes of Infantile Cholestasis and Their Correlation With Outcome: A Multicenter Study. Am J Surg Pathol. 2016 Dec;40(12):1601–15.
- Shneider BL, Moore J, Kerkar N, Magee JC, Ye W, Karpen SJ, et al. Initial assessment of the infant with neonatal cholestasis—Is this biliary atresia? Alpini GD, editor. PLOS ONE. 2017 May 11;12(5):e0176275.