
Interesting Case October 2021
Case contributed by:
Hao Wu, MD
Assistant Professor, Department of Pathology, Yale School of Medicine
Have an interesting case to share?
Contact HPHS newsletter committee chair Sadhna.Dhingra@ProPath.com
Case history
A 40- year-old fitness trainer presented with persistant hyperlipidemia and elevated liver biochemistry, with ALT at 200 U/L, and AST 140 U/L. ALP, GGT and bilirubin were within normal limits. The patient had been on atorvastatin for 2 years without improvement in hyperlipidemia. There is family history of coronary artery disease. He denied smoking, drinking, or taking supplement.
On CT, there was diffuse fat signal in the liver with mild hepatomegaly.
Figure 1. H&E.
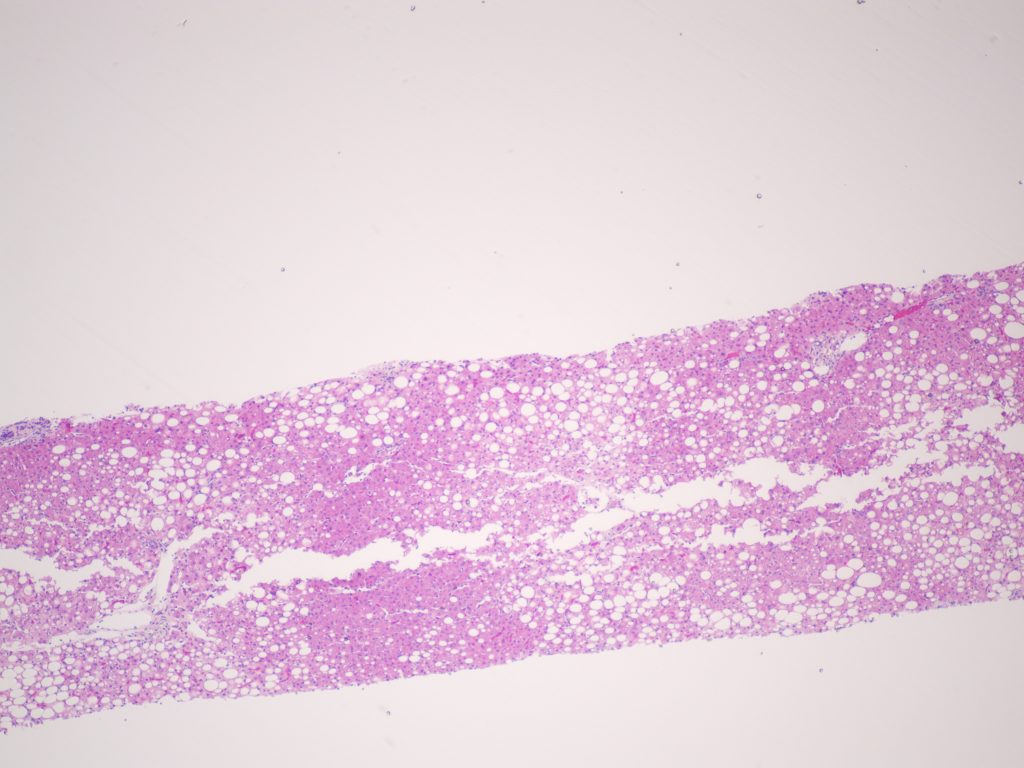
Figure 2. H&E.
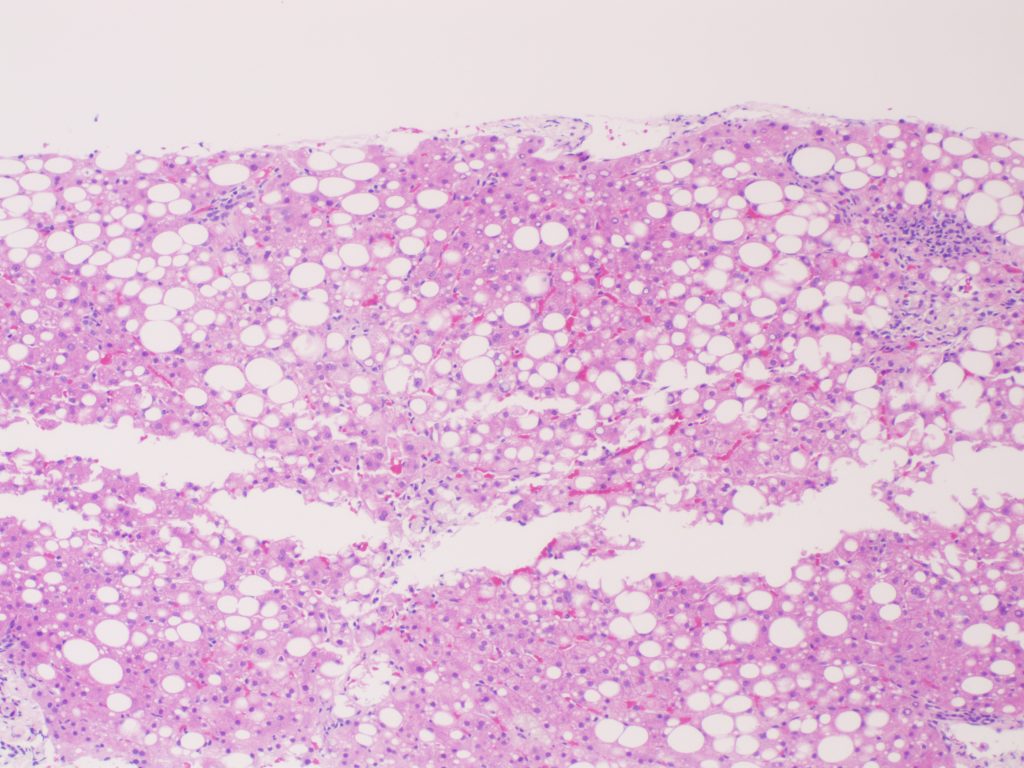
Figure 3. H&E.
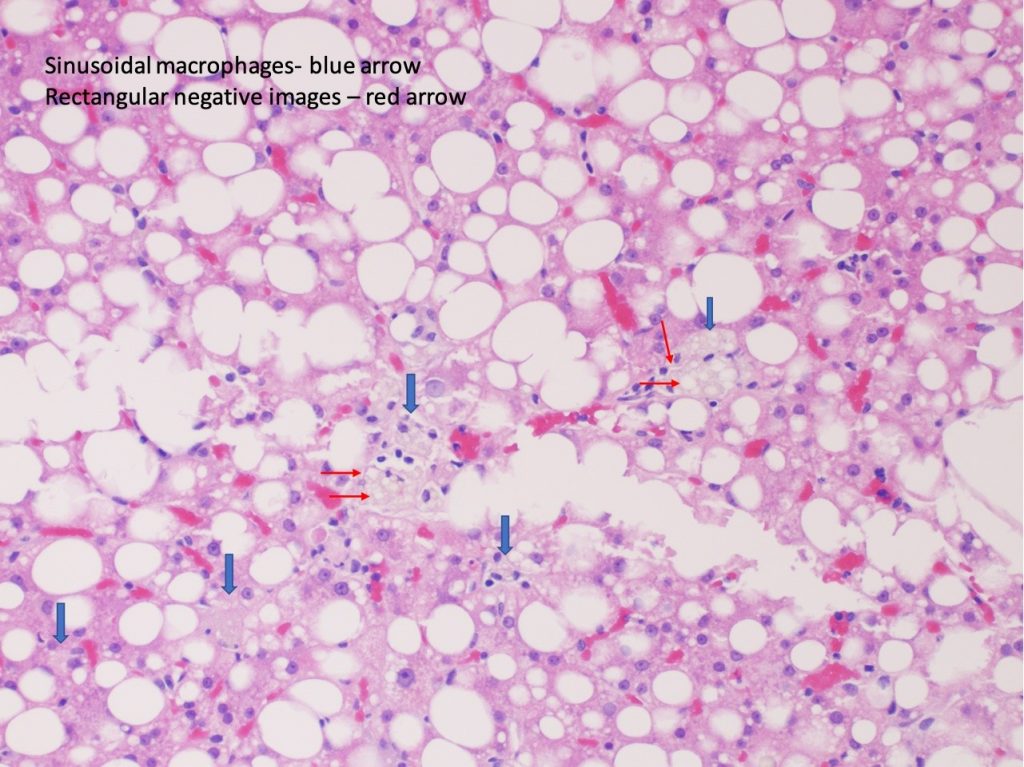
Figure 4. PAS-D.
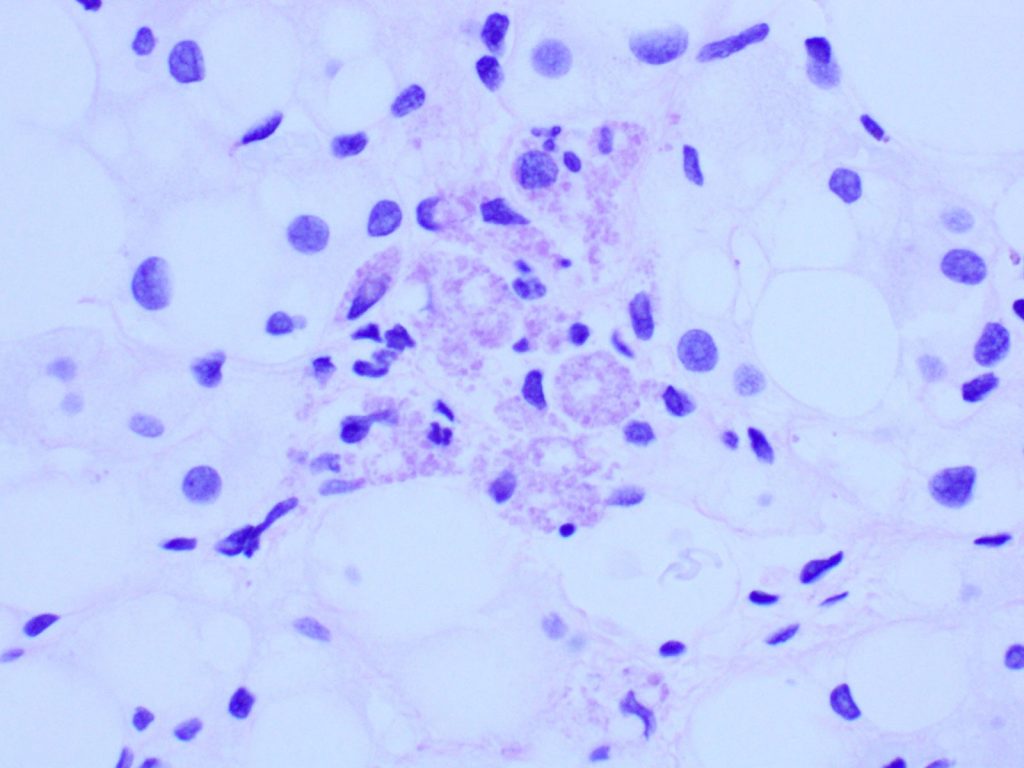
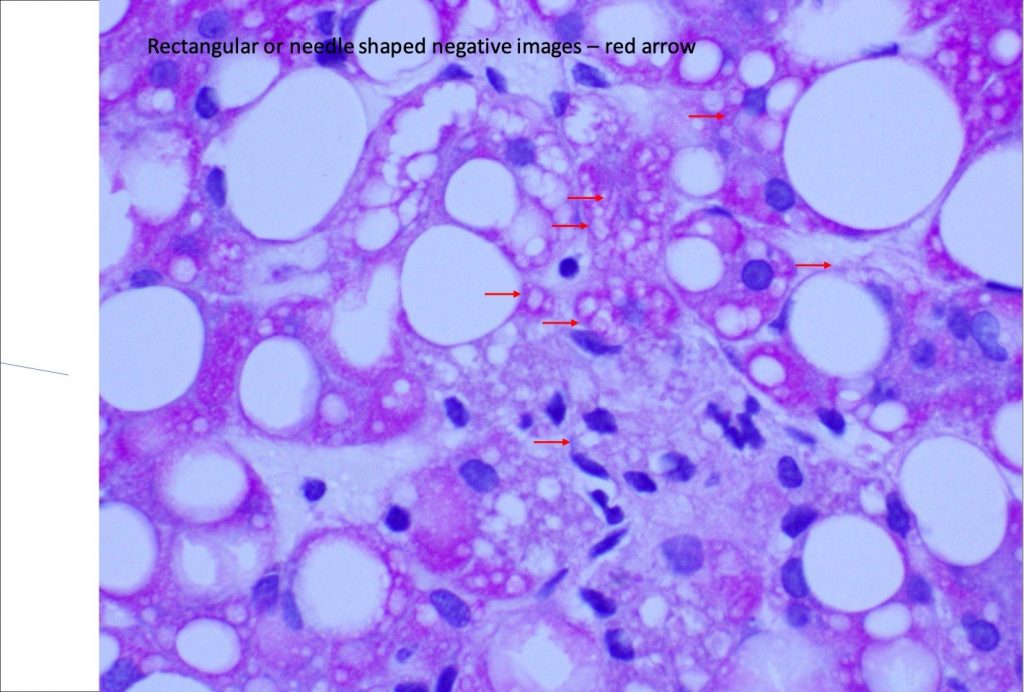
Figure 5. PAS.
Pathologic findings
Liver biopsy revealed macrovesicular steatosis involving about 50% liver parenchyma (Fig 1 and 2). No ballooning degeneration was seen. Rare lymphocytic lobulitis was present, focally up to 2 foci per 20x field. No portal inflammation was present. There were prominent ceroid laden in sinusoidal macrophages (Fig 3) and multiple lipogranulomas. The ceroid pigment was better appreciated on PAS/D stain (Fig 4). The lipogranulomas were predominantly located in zone 2 and zone 3. No hepatocytic ceroid pigment was seen. Higher magnification of PAS-stained slides showed geographic negative spaces within the kupffer cells (Fig 5), suggestive of intracellular crystals.
Presence of steatosis in the liver biopsy of a lean patient with hepatomegaly, and additional histological features of ceroid laden macrophages along with possible intracellular crystals in the macrophages prompted a blood lysosomal acid lipase quantification and LIPA gene sequencing. Blood lysosomal acid lipase levels were below reference levels (performed in two separate reference laboratories). Genetic work up revealed a heterozygous mutation in LIPA gene.
Diagnosis
Cholesteryl ester storage disease (lysosomal acid lipase deficiency).
Discussion
Lysosomal acid lipase deficiency (LAL-D) is a rare autosomal recessive genetic disorder caused by mutations of lysosomal acid lipase gene (LIPA), located on chromosome 10q23.2 that encodes for the enzyme lysosomal acid lipase (LAL). Lysosomal acid lipase is a key enzyme in lipid metabolism and is responsible for hydrolysing the cholesteryl esters and triglycerides into free cholesterol and fatty acids. Genetic mutations cause absent or variably decreased LAL levels which leads to accumulation of cholesterol esters and triglycerides in many tissues.
LAL-D has two main clinical phenotypes: early-onset Wolman disease (WD) and late onset cholesterol ester storage disease (CESD) (1). Wolman disease is characterized by complete absence of LAL that leads to triglyceride and cholesterol ester accumulation within the reticuloendothelial cells. It typically manifests in the first few weeks of life with malabsorption, hepatosplenomegaly, failure to thrive, adrenal insufficiency and multi-organ failure. The life expectancy is one year without treatment because of bone marrow and liver failure. In contrast, CESD is a less severe form of LAL deficiency which is characterized by partial presence of LAL levels. Depending on the particular types of mutation, the disease presentation can be quite variable, with most reported cases discovered in childhood and less frequently in adulthood. The clinical manifestations include mild hepatomegaly, microvesicular steatosis, splenomegaly, accelerated atherosclerosis and dyslipidemia (2).
CESD is pan-ethnic and disease incidence is unknown (3). Mild form of CESD, as discussed in this case, may remain under-recognized or misdiagnosed. The natural history and clinical manifestations of disease in adults are less well-defined and the diagnosis is often incidental (4). The clinical phenotype depends on the residual enzyme activity. With the recent approval of enzyme replacement therapy (sebelipase alfa), an accurate diagnosis can offer great benefits to patients to decrease cardiovascular risks and liver damages. The diagnosis can be established with help of liver biopsy findings (3).
Role of liver biopsy in LAL-D
All patients with LIPA gene mutation have hepatic steatosis due to intracellular accumulation of cholesteryl esters and triglycerides in the lysosomes. However, hepatic steatosis has diverse etiologies. The most common etiology is metabolic syndrome in which the triglycerides are cytosolic in distribution. It is difficult to differentiate hepatic steatosis of LAL-D from that of metabolic syndrome in a routine hematoxylin and eosin stained section of formalin fixed paraffin embedded (FFPE) liver biopsy. In LAL-D patients, the characteristic steatosis described in literature is a microvesicular steatosis with minimal zonal differences (5). However, foci of microvesicular steatosis have also been described in fatty livers of metabolic syndrome. The biopsy in our case did not show the characteristic microvesicular steatosis. Immunohistochemical staining with lysosomal markers such as luminal cathepsin D, membrane lysosomal markers, LAMP 1 and LAMP 2 and lysosomal integral membrane protein 2 (LIMP 2) can help identify the lysosomal lipid distribution. It appears as a positive staining around the lipid vacuoles.
Other histological feaures that can point to LAL-D include infiltration of sinusoids and portal tracts by ceroid laden macrophages and presence of cholesterol ester crystals. The ceroid pigment in the macrophages is easily recognized on PASD stain. Hepatocytes in LAL-D lack ceroid accumulation or lipofuscin pigment (5). Cholesterol ester crytals in hepatocytes and kupffer cells are pathognomic for LAL-D. These are best identified in fresh frozen tissue when observed under polarized light as birefringent crystals in Maltese cross pattern. Formalin fixation and subsequent dehydration steps of tissue processing leads to extraction of the lipid crystals leaving behind rhomboid or plate like clear spaces/clefts.
In general, NAFLD-related dyslipidemia responds to life style modification and statin therapy. Normal body mass index, persistent hyperlipidemia on statins and negative images of cholesteryl ester crystals are all helpful clues to help the patients avoid a liver transplantation.
Learning points:
It is important to determine the role of LAL deficiency in NAFLD. Metabolic syndrome related NAFLD and dyslipidemia responds to life style modification and lipid lowering drugs. Whereas, steatosis/steatohepatitis, its progression to cirrhosis and dyslipidemia in a LAL-D patient can respond to enzyme replacement therapy. The case illustrated here demonstrates a clinical and histopathological scenario that should trigger a clinical consideration of LAL deficiency to be confirmed or excluded by blood work-up and genetic testing.
Clinical features that lead to suspicion of LAL-D include: lean patients with hepatomegaly, elevated transaminases and dyslipidemia (high LDL-C and low HDL-C)
Histopathological features observed in LAL-D patients include: hepatic steatosis (particularly microvesicular), infiltration by ceroid laden macrophages, presence of birefringent cholesterol ester crystals on fresh frozen sections or remnants clefts on FFPE tissue and absence of lipofuscin in hepatocytes. Immunostaining with lysosomal markers can confirm the lysosomal distribution of lipid accumulation.
References
- Pericleous M, Kelly C, Wang T, et al: Wolman’s disease and cholesteryl ester storage disorder: the phenotypic spectrum of lysosomal acid lipase deficiency. Lancet Gastroenterol Hepatol. 2017;2(9):670-679.
- Burton BK, Deegan PB, Enns GM et al. Clinical features of lysosomal acid lipase deficiency – a longitudinal assessment of 48 children and adults. J Pediatr Gastroenterol Nutri, 2015; 61: 619-625.
- Bernstein DL, Hulkova H, Bialer MG, Desnick RJ: Cholesteryl ester storage disease: Review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58:1230-1243
- Baratta F, Pastori D, Polimeni L, Tozzi G, Violi F, Angelico F, Del Ben M. Does Lysosomial Acid Lipase Reduction Play a Role in Adult Non-Alcoholic Fatty Liver Disease? Int J Mol Sci. 2015 Nov 25;16(12):28014-21.
- Hůlková H, Elleder M. Distinctive histopathological features that support a diagnosis of cholesterol ester storage disease in liver biopsy specimens. Histopathology. 2012 Jun;60(7):1107-13. doi: 10.1111/j.1365-2559.2011.04164.x. PMID: 22621222.