
Interesting Case January 2020
Case Contributed by:
Oscar W. Cummings, M.D.
IU Health Department of Pathology and Laboratory Medicine
Indianapolis, IN
Have an interesting case to share?
Contact HPHS newsletter committee chair Sadhna.Dhingra@ProPath.com
Clinical History
The patient is a 57-year-old female who presented with fatigue and back pain. An initial examination revealed an elevated serum ferritin level and mild anemia. She was referred to a hepatologist who under took a more extensive workup. The patient worked as a photographer and was otherwise well. She had 2 children, no history of transfusions and did not drink alcohol. Medications were limited to nutritional supplements. There were no physical findings. An MRI showed numerous hepatic cysts. FibroScan had a CSM of 3.6 kPa, F0, and a CAP score of 222 dB/m. Her LFTs (AST, ALT, ALP, bili) were within normal limits. Other findings included: Hemoglobin 11.6 gm/dl (12-15), HCT 34.9% (35-49%), serum iron 44mCg/dl (50-212), ferritin 998.5 ng/dl (10-106), % saturation 13% (15-55), total iron binding capacity 338 mCg/dl (250-400). HFE analysis was normal. Her serum copper levels were low, < 10 mCg/ dl (80-155) and ceruloplasmin was undetectable < 2.0 mg/dl (14-57). A 24 hour urine copper was negative for copper with adequate controls. Other findings included: glucose 94 mg/dl, Hg1Ac 5.9 % (4-5.6%). Autoimmune markers (AN A, ASMA, L KM and AMA) were all within normal limits as was Alpha-1 antitrypsin levels. Hepatitis virus studies (HAV, HBV, HCV, HEV) were negative. A liver biopsy was performed and sent with a notation to rule out Wilson disease, perform copper stains and analysis.
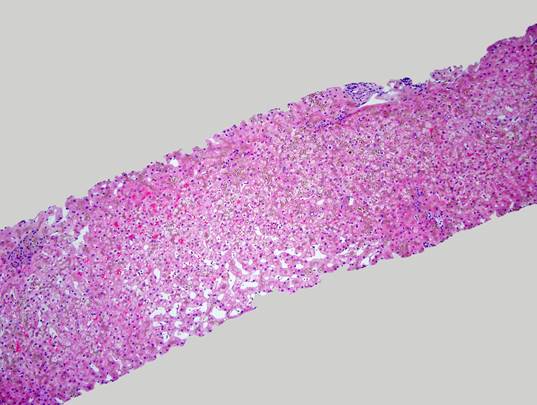
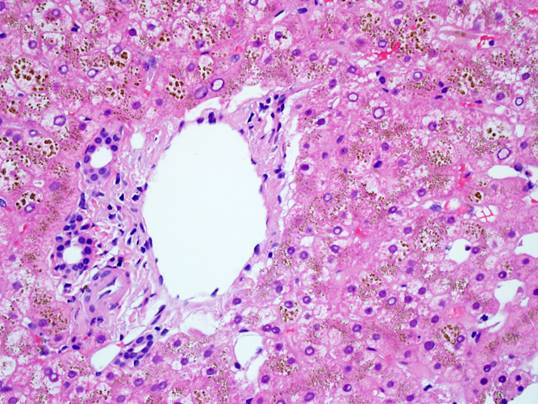
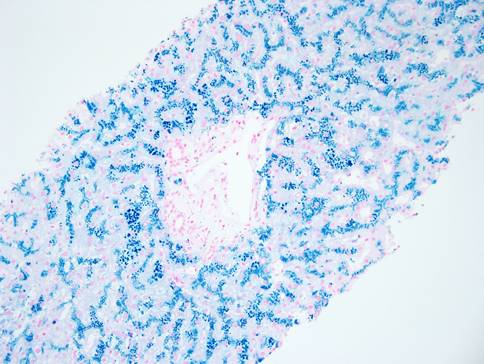
Answer:
Hemochromatosis, grade 4, secondary to aceruloplasminemia
Discussion
The liver biopsy shows an intact lobular architecture. Portal tracts including the intralobular bile ducts are essentially unremarkable. The hepatocytes show a vague tan appearance at low power and there is some sinusoidal dilation. High power shows an unremarkable portal tract with tan refractile granules in the cytoplasm of the surrounding hepatocytes. A gross photograph of the iron stain slide shows visible discoloration of the tissue without magnification – grade 4 hemochromatosis. The iron stain itself shows diffuse hepatocellular iron deposition with a pericanalicular pattern. There was no fibrosis – trichrome stain not shown.
Hemochromatosis is one of the more common hereditary disorders. There are multiple molecules involved in iron hemostasis and disruption of any of them can lead to iron accumulation in the patient. The most well-known cause of genetic hemochromatosis is due to mutations in the HFE gene; however, that was clearly not a factor in this patient since those studies were normal. The constellation of mild anemia, high serum transferrin and low to absent ceruloplasmin – as seen in this patient – are very characteristic of aceruloplasminemia associated hemochromatosis.
The first case of aceruloplasminemia (ACP) was reported in 1987 and there were approximately 50 case reports in the literature through 2010. Most of the original reports came from Japan but ACP has also been reported in many other parts of the world. The patients often present between the ages of 30 and 70 years with a mean age of 50. In addition to the anemia and elevated transferrin, they typically exhibit retinal degeneration, diabetes mellitus, neurologic disorders (blepharospasm, tremors, chorea, ataxia) and cognitive dysfunction including apathy and forgetfulness. The anemia and diabetes tend to occur early in the course of disease while the neurologic manifestations appear later. The ceruloplasmin (CP) gene resides on chromosome 3 (3q24-q25) and has 20 exons. The normal gene product is a single polypeptide of 1,046 amino acids that can bind up to 6 atoms of copper. There are 2 distinct isoforms produced by alternate splicing of the last 2 exons. The soluble form is produced almost exclusively in the liver and is the major (95%) carrier protein for copper in the serum. The other isoforms contains a membrane binding site and is produced in multiple cells including hepatocytes, astrocytes, macrophages, pancreatic cells and retinal epithelial cells. This isoforms closely interacts with the Ferroportin molecule regulating iron passage across the cell membrane. As the intracellular Fe2+ passes through the pore, CP enzymatically converts it to Fe3+ which can now bind to transferrin. The Fe3+transferrin complex can now bind to the transferrin receptors (TfR1) on multiple cells including erythroid precursors and astrocytes providing iron for normal cell function. ACP is inherited in an autosomal recessive manner. Multiple different mutations have been described – 28 missense, 17 frame shift, 13 splicing and 8 nonsense – leading to a non-functional gene product. Mutations have been reported in all the exons except for numbers 8,19,20. Without CP, there is no Fe2+ to Fe3+ conversion so Fe 2+ iron is trapped in cells leading to iron overload and cytotoxicity. The mechanism of serum transferrin elevation is unclear but it may be compensatory overproduction in response to lack of Fe3+ iron.
The pathologic distribution of iron overload in ACP is relatively well catalogued. In the liver, there is dense relatively pan-acinar accumulation of iron preferentially in a canalicular distribution without significant fibrosis. In the pancreas, iron can accumulate in both acinar cell epithelium and endocrine cells possibly leading to the observed diabetes. In the brain, iron overload is most common in the globus pallidus followed by the putamen, cerebral cortex and cerebellar cortex. Marked iron overload and neuronal loss are common in the basal ganglia while there is often frontal cortex sparing. The iron deposition is more prominent in astrocytes than in neurons. The extent of iron deposition in the heart is variable. It is uncommon in the Japanese cohort but multiple cases of cardiomyopathy have been reported in other countries.
In the illustrated case, the patient clearly has marked hepatic iron overload. While her glucose values were within normal limits, the hemoglobin A1c suggests a prediabetic state. The sinusoidal dilation in the liver biopsy raises the possibility of mild congestive hepatopathy so a cardiac scan is warranted to evaluate the possibility of hemochromatosis associated cardiomyopathy. It is possible that therapy may improve some of these end organ derangements. However it is most important to instigate appropriate therapy so as to avoid development of the neurologic sequelae. Iron chelating agents (desferrioxamine, deferiprone, deferasirox) are the main stays of therapy along with supportive measures including fresh frozen plasma, vitamin E and zinc. Some studies have shown that these therapies may improve and/or prevent neurologic damage.
Take home points
1. Patients with Wilson disease generally do not present in their 50s
Wilson disease is an autosomal recessive copper storage disorder caused by mutations in the Wilson’s disease gene (ATP 7B). Patients usually present in childhood or young adulthood with hepatic or neurologic dysfunction. There are a handful of case reports of Wilson disease presenting in patients over 50, but this late presentation of Wilson disease is less common than in ACP. The Wilson gene product is an intracellular transmembrane copper transporter that is key in incorporating copper into ceruloplasmin and in moving copper out of the hepatocyte into bile. Mutations in ATP7B disrupt these functions leading to toxic accumulation of copper in the cells. In Wilson’s disease, the CP gene product is normal. However, without the necessary copper ions the protein is unstable and serum levels are low but functional. In general, Wilson’s disease patients do not accumulate iron.
2. Special stains requested by the clinician may not be helpful
We did not perform the requested copper stain since the patient did not have Wilson disease based on the clinical information provided. Further, copper stain is often negative in patients with Wilson disease especially in children with fulminant presentations. In those settings, the best diagnostic tests are tissue copper analysis and 24 hour urine copper levels, both of which would be significantly elevated in Wilson disease. To address the clinician’s concerns, we performed metal analysis of the liver biopsy: tissue iron 27,987.8 ug/g (ref 200-1,600), tissue copper 31.1 ug/g (ref 15 -55). The markedly elevated tissue iron levels and normal tissue copper levels support the diagnosis of ACP.
3. The clinical information is very important in making an anatomic surgical pathology diagnosis
We as pathologist know this adage well but it is often lost on our clinical colleagues. If no clinical information had been provided with this case, we would have rendered the correct diagnosis of hemochromatosis but it would have made no sense to our clinical colleagues. With the clinical history we were able to make a very specific diagnosis that had not been considered previously by the clinicians. Hopefully, this will lead to timely therapy which will spare the patient from developing neurologic sequela.
References
Miyajima H, Hosoi Y. Aceruloplasminemia. 2003 Aug 12 [Updated 2018 Sep 27]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.
Kaler SG. Inborn errors of copper metabolism. Handb Clin Neurol. 2013 ; 113: 1745–1754.
Marchi G, Busti F, Zindanes AL, Castagna A, Girelli D. Aceruloplasminemia: A Severe Neurodegenerative Disorder Deserving an Early Diagnosis. Front Neurosci 2019, 13:325. doi: 10.3389
Pietrangelo A, Caleffi A, Corradini E. Non-HFE Hepatic Iron Overload. Semin Liver Dis 2011;31:302–318.
Pietrangelo A. Genetics, Genetic Testing, and Management of Hemochromatosis: 15 Years since Hepcidin. Gastroenterology 2015;149:1240–1251
Weiss KH. Wilson Disease. 1999 Oct 22 [Updated 2016 Jul 29]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.