
Interesting Case April 2016
Clinical History
A 65 year old woman was found to have abnormal liver tests during a follow up visit for monoclonal gammopathy of uncertain significance (MGUS), IgG kappa. High transaminase levels prompted a needle core biopsy of the liver.
Lab Values
ALT and AST >200 U/L
Antimitochondrial antibody (AMA) was negative.
Photomicrographs
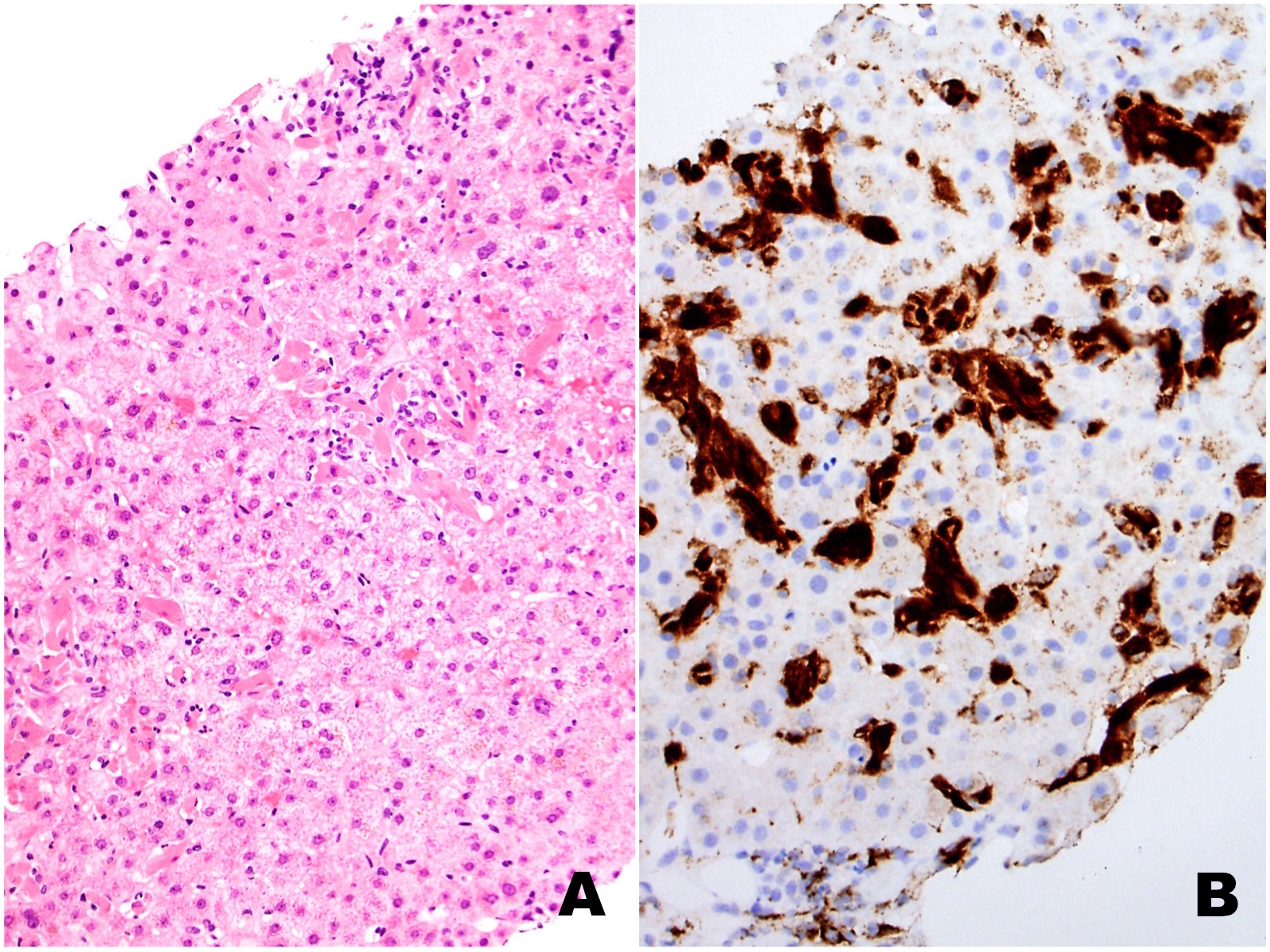 |
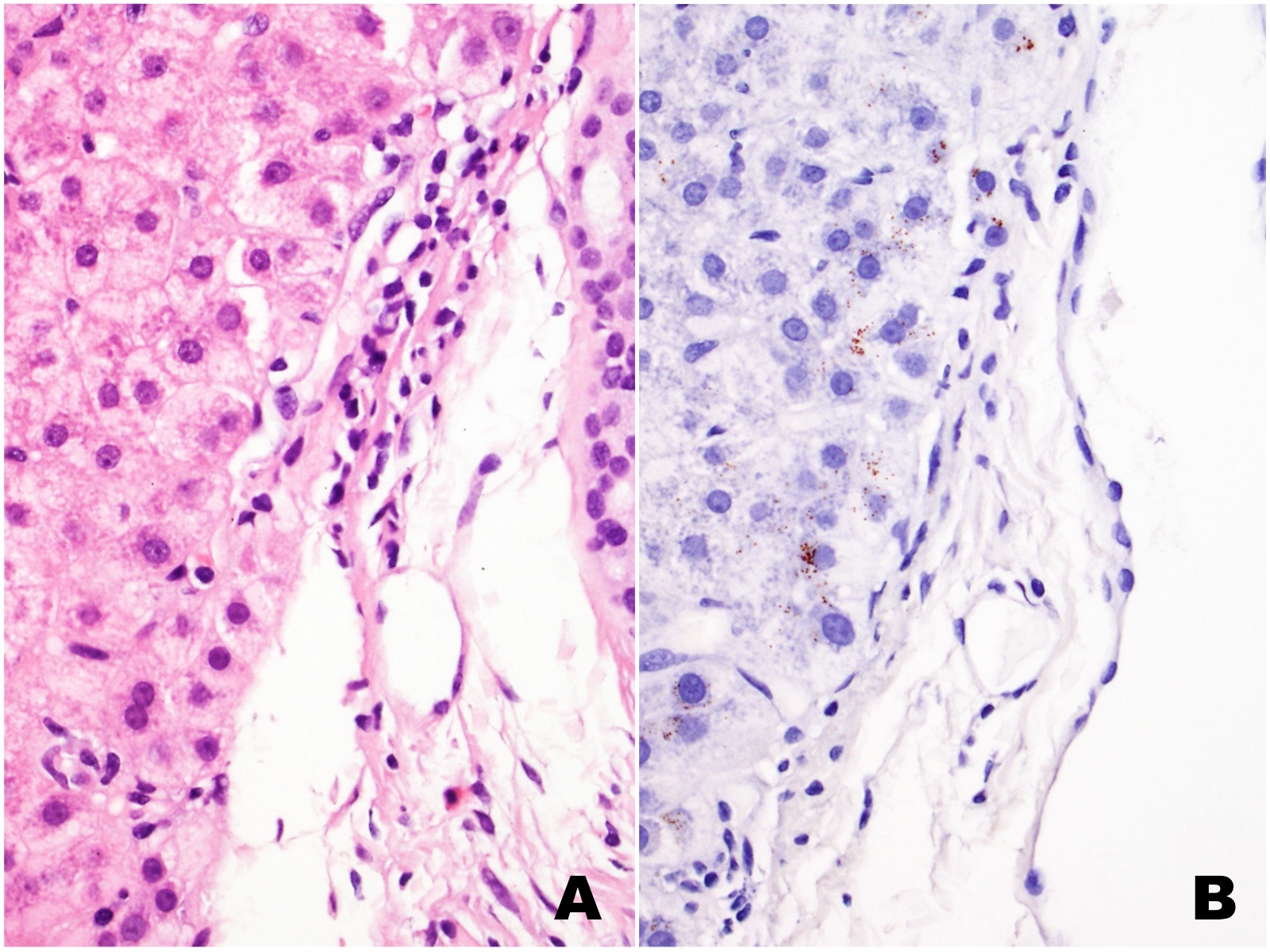
| Figures 1A (H&E) and 1B (CD68) |
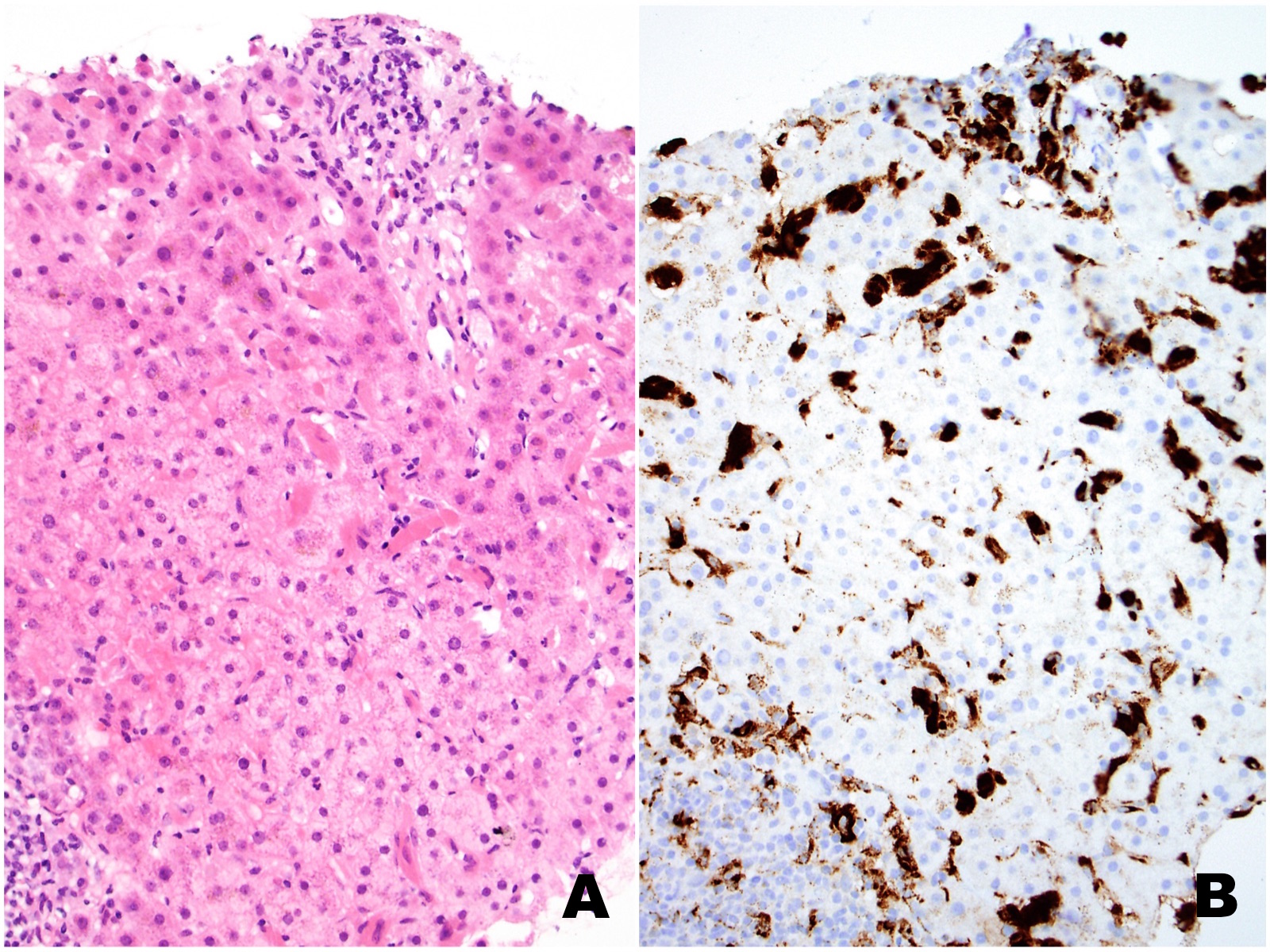 |
| Figures 2A (H&E) and 2B (CD68) |
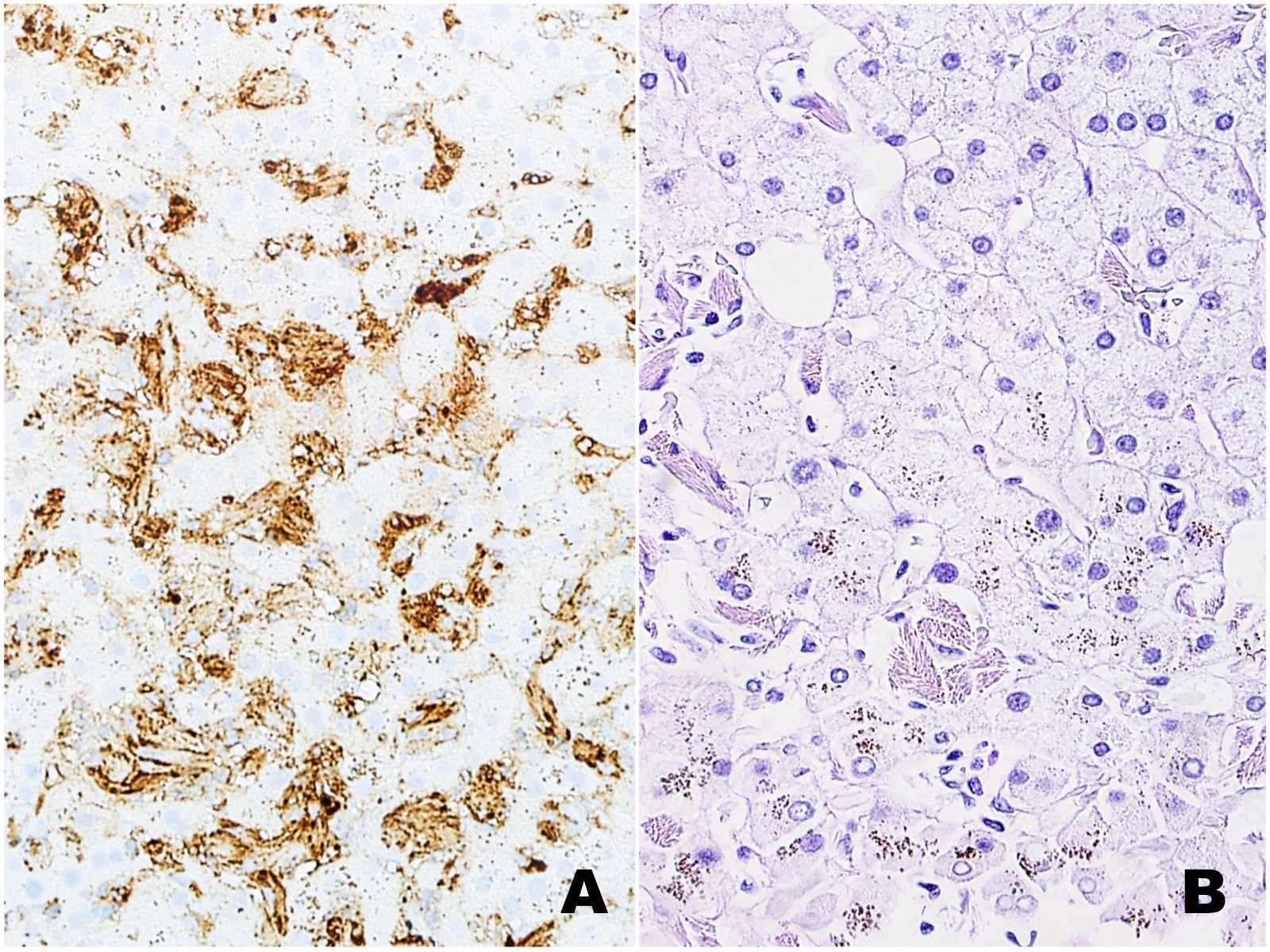 |
| Figures 3A (Ig kappa) and 3B (PAS-D) |
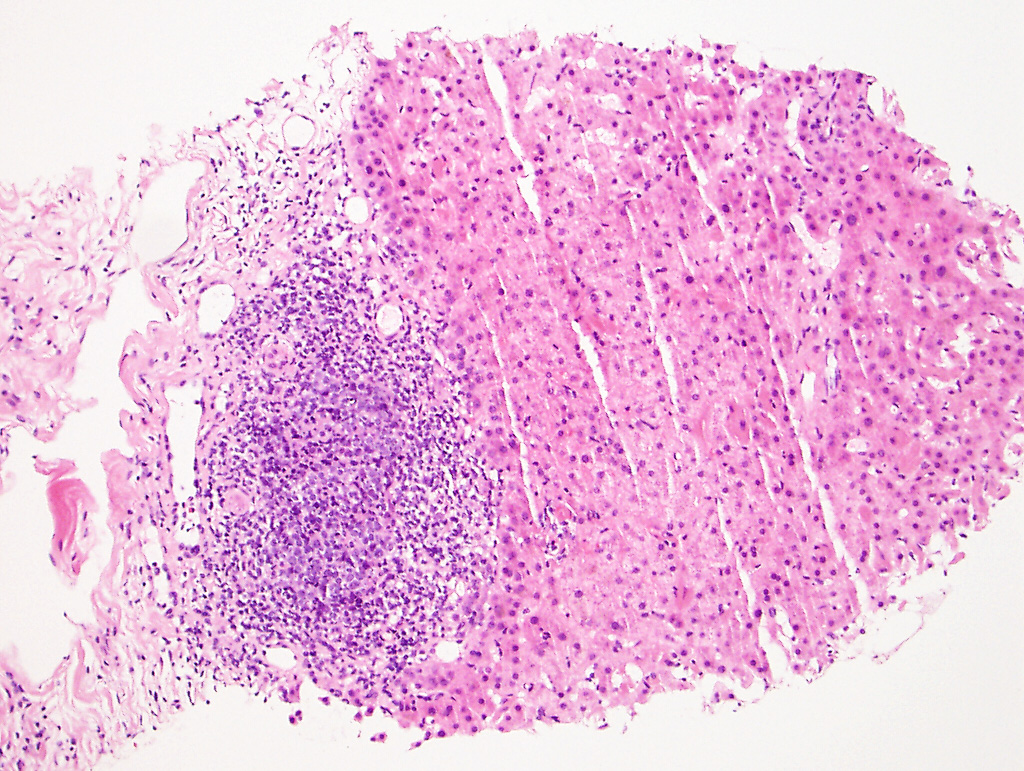 |
| Figure 4 (H&E) |
 |
| Figures 5A (H&E) and 5B (Rhodanine) |
Liver Biopsy Findings
The liver biopsy shows extensive sinusoidal infiltration by bland appearing plump eosinophilic cells with somewhat retractile or granular cytoplasm (Figures 1A and 2A). These cells are positive for CD68 confirming they are histiocytes (Figures 1B and 2B). The histiocytes contain crystalline immunoglobulin kappa highlighted on the Ig kappa and PAS-D stains (Figures 3A and 3B, respectively). Ig lambda was negative.
In a single portal tract there is mild inflammation associated with bile duct injury (Figure 4). One periportal area is positive for focal copper accumulation on the Rhodanine stain (Figures 5A and 5B, respectively). There were no histologic features of a concomitant clonal plasma cell or lymphoproliferative process.
Diagnosis
Crystal-storing histiocytosis (immunoglobulin variant).
Focal mild portal inflammation with associated bile duct injury.
Major Learning Points
• Crystal-storing histiocytosis (CSH) is a subtle histologic diagnosis when it involves the liver.
• A systematic approach to liver biopsy evaluation will help avoid missing sinusoidal infiltrates.
• The published literature on CSH reveals that most cases are due to plasma cell proliferative disorders (such as multiple myeloma and MGUS) with rare cases due to other causes such as i) Charcot Leyden crystals, ii) clofazimine, iii) marked polyclonal plasma cell rich inflammation and iv) hereditary cystinosis.
• It is important to evaluate the biopsy which shows CSH for a simultaneous clonal lymphoproliferative or plasma cell proliferative process. If an initial work up is negative, the patient should have periodic follow up as the diagnosis of CSH can precede the diagnosis of a plasma cell proliferative disorder.
Discussion
Crystal-storing histiocytosis (CSH) is rare. In the liver, the diagnosis is subtle because the sinusoidal location of eosinophilic histiocytes blends in with the adjacent eosinophilic hepatocytes. CSH can be due to a number of causes, but >90% of cases are due to plasma cell proliferative disorders including multiple myeloma, lymphoplasmacytic lymphoma and MGUS. Often times these underlying disorders are known to the clinical service, however, it is useful to include this association in the pathology report for clinicians who may not be aware. Furthermore, in situations in which a plasma cell proliferative disorder has not yet been established, pointing out this association in the pathology report may facilitate further clinical evaluation. It is also worth noting that a recently characterized subset of cases of CSH may be due to clofazimine, Charcot-Leyden crystals, rheumatoid arthritis, marked polyclonal plasma cell inflammation or hereditary cystinosis. These should also be included in the differential diagnosis for cases where no history is present.
CSH typically presents as either a generalized disease with multi-organ involvement or a localized process. The liver is involved in about half the cases of generalized disease. There are no recognized histologic differences between localized and generalized cases. This distinction is made based on clinical information and review of specimens from multiple sites.
Histologically, cases of CSH are characterized by an abundance of eosinophilic histiocytes with plump cytoplasm containing birefringent crystalline material. In the liver, the histiocytes most often show a sinusoidal pattern of distribution, but mass-like lesions can occur. The histiocytes show the typical immunophenotype with CD68 and CD163 expression. If the crystals are composed of immunoglobulin, they may be clonal or polyclonal and composed of light chains or rarely heavy chains. Immunoglobulin crystals are often needle shaped but in the authors’ experience, other shapes can be seen. The mechanism for crystal formation is poorly understood.
CSH should be distinguished from Langerhans cell histiocytosis, Gaucher’s disease and other lysosomal storage diseases and infection. Langerhans cell histiocytosis is often accompanied by a prominent eosinophilic infiltrate and the neoplastic cells display grooved nuclei. Lysosomal storage diseases are characterized by foamy rather than birefringent cytoplasm and immunoglobulin immunohistochemistry is negative. Electron microscopy and enzyme assays may be helpful in confirming and refining the diagnosis of a lysosomal storage disease. Finally, infections can be excluded based on histochemical stains (such as AFB and GMS) together with clinical information and blood tests.
This case is unusual in that there is focal bile duct injury with associated periportal copper accumulation. The very focal nature of these findings is insufficient for a diagnosis of primary biliary cirrhosis and the patient’s AMA is negative.
In conclusion, CSH is a rare, histologically subtle disorder which may affect the liver. Importantly, most cases are due to an underlying plasma cell proliferative disorder.
This case was contributed by:
Rondell P Graham, MBBS, Gastrointestinal/Liver Pathology Fellow, Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN
and
Vashal Chandan, MBBS, Assistant Professor of Pathology, Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN
Reference:
S Dogan et al. Head and Neck Pathol 2012;6:111-120.